前言
- 工具:Rstudio+BioinfoJupyter
- 环境:我院深度学习工作站
下载数据
使用GEOquery包可直接从GEO下载芯片原始数据:
library(GEOquery)
getGEOSuppFiles("GSE33672")
下载完毕后会得到GSE33672文件夹,将其中的.tar文件解压,得到四个压缩后的.CEL.gz文件;将其全部解压至data文件夹。
读取数据
该芯片建议使用oligo包读取。
library(oligo)
data.dir <- "data/"
(celfiles <- list.files(data.dir, "\\.CEL$"))
data.raw <- read.celfiles(filenames = file.path(data.dir, celfiles))
读入成功,输出以下内容:
Platform design info loaded.
Reading in : data//GSM832614_MX2-1.CEL
Reading in : data//GSM832615_MX2-3.CEL
Reading in : data//GSM832616_MXEV2.CEL
Reading in : data//GSM832617_MXEV3.CEL
使用rma算法标准化:
data.eset <- rma(data.raw)
data.exprs <- exprs(data.eset)
标准化成功,输出以下内容:
Background correcting
Normalizing
Calculating Expression
对样本名称进行简化:
treats <- strsplit("MX MX MXEV MXEV", " ")[[1]]
(snames <- paste(treats, 1:2, sep = ""))
sampleNames(data.raw) <- snames
pData(data.raw)$index <- treats
sampleNames(data.raw)
pData(data.raw)
- ‘MX1’
- ‘MX2’
- ‘MXEV1’
- ‘MXEV2’
- ‘MX1’
- ‘MX2’
- ‘MXEV1’
- ‘MXEV2’
| index | |
|---|---|
| MX1 | MX |
| MX2 | MX |
| MXEV1 | MXEV |
| MXEV2 | MXEV |
质量控制
首先查看是否存在缺失值:
sum(is.na(exprs(data.raw)))
0
绘制箱型图,检查标准化效果:
par(mfrow=c(1,2))
boxplot(exprs(data.raw), names = NA, col = as.factor(treats))
legend("topright", legend = unique(treats), fill = as.factor(unique(treats)),
box.col = NA, bg = "white", inset = 0.01)
boxplot(exprs(data.eset), names = NA, col = as.factor(treats))
legend("topright", legend = unique(treats), fill = as.factor(unique(treats)),
box.col = NA, bg = "white", inset = 0.01)
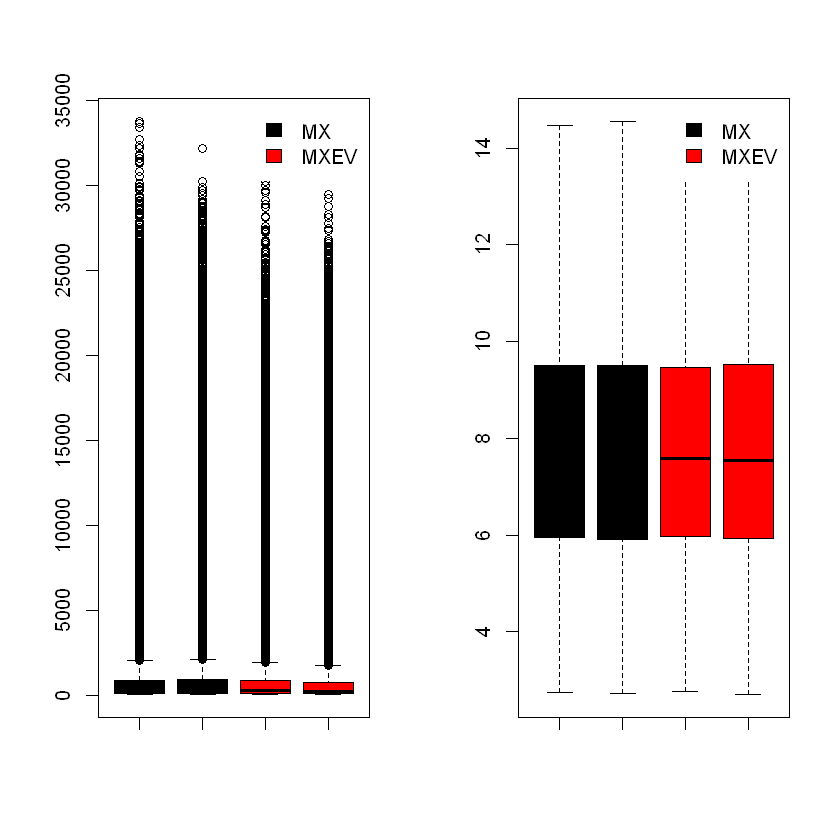
对比可知数据得到有效集中,标准化取得了良好的效果。
绘制MA图,查看标准化后各芯片中M、A关系:
par(mfrow = c(2, 2))
MAplot(data.raw[, 1:4], pairs = FALSE)
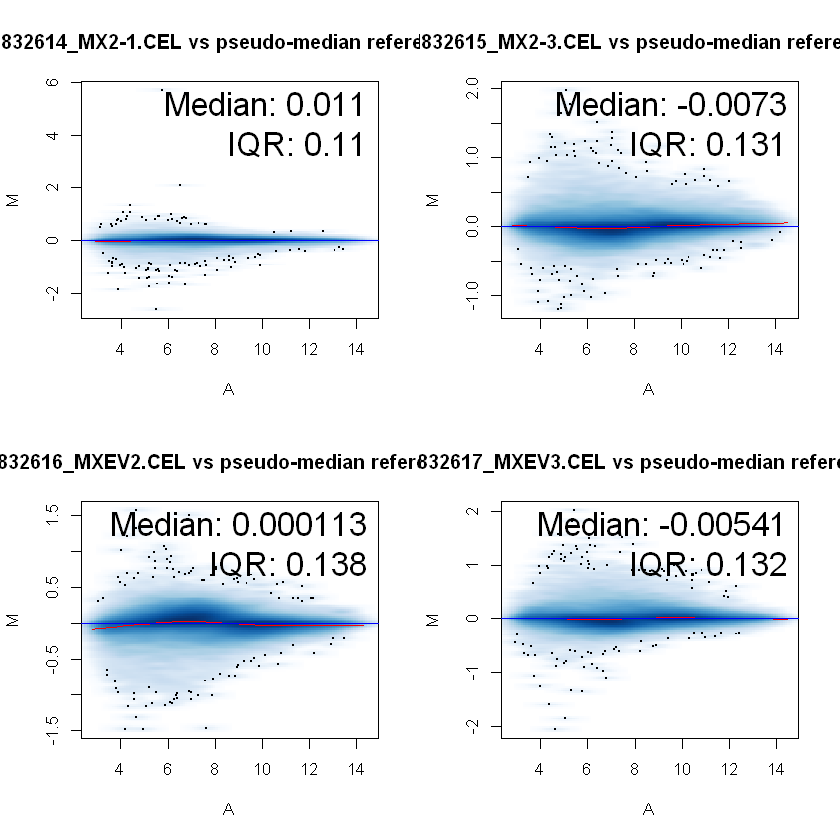
中值均贴近零线,与箱型图结果相符,质量过关。
数据清洗
筛去p值<0.05的样本:
pinfo <- getProbeInfo(data.raw)
xpa <- paCalls(data.raw)
AP <- apply(xpa, 1, function(x) any(x < 0.05))
xids <- as.numeric(names(AP[AP]))
fids <- pinfo[pinfo$fid %in% xids, 2]#id转化
data.exprs2 <- data.exprs[rownames(data.exprs) %in% fids, ]
清洗成功,输出:
Computing DABG calls...
OK
MA图检查结果:
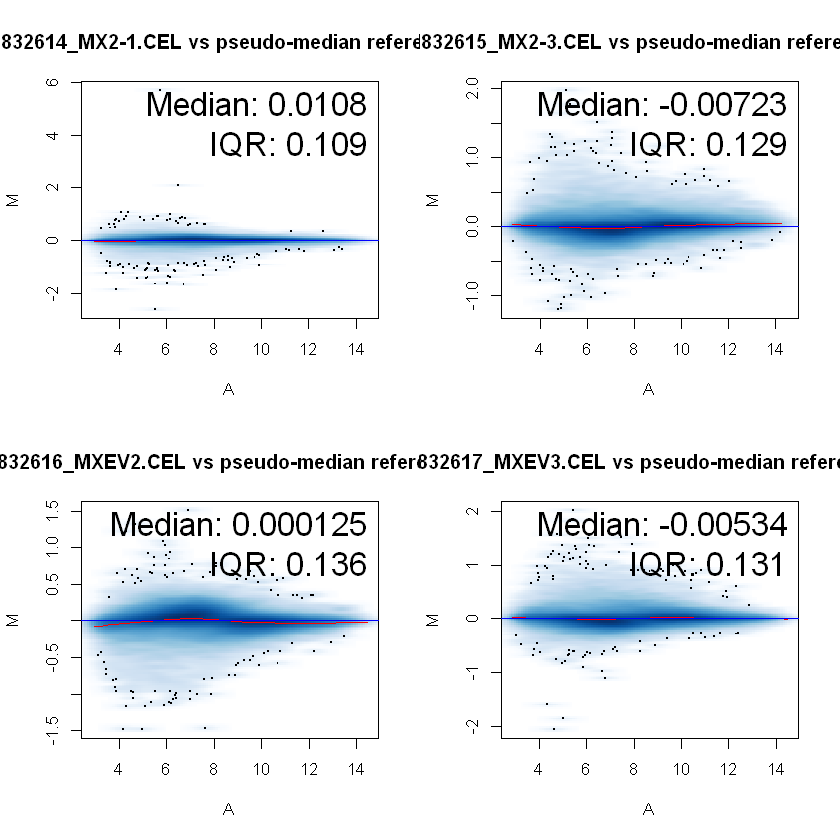
对比可知中值与IQR进一步收敛。最终得到表达矩阵data.exprs2。
附录
R笔记本:点击此处